1 November 2025 by Remco Bouckaert
Most BEAST analyses involve alignments of nucleotide or amino acid sequences. However, when there is ample prior information available about the structure of the phylogeny, it is still possible to get useful information from a Bayesian phylogenetic analysis. To demonstrate this, we have a look at the analysis of global religions (Ejova et al, 2025).
Using encyclopaedias and other sources, a database was created of a wide range of religious denominations belonging to the Big Five world religions and their relatives, along with their genealogical relationships. These relationships were encoded as most recent common ancestor (MRCA) constraints, both in topology and in time. The tree is not completely resolved and there remained two kinds of uncertainty:
- Not all relationships could be unambiguously determined – sometimes it was unclear from historical documents whether a religion was part of a clade or not. For that reason, the
MRCAPriorWithRogueswas introduced in the BEASTLabs package. Essentially, it enforces a clade to be monophyletic after removing a specified set of rogue taxa have been removed from the tree. - There is uncertainty about the timing, especially of splits in the tree farther in the past. These were encoded as node calibrations in the analysis.
Together, this allowed inferring posteriors on the three big trees: one for Judeo-Christianity, one for Islamic and one for Indo-Iranian, which includes Buddhism and Hinduism. Since there is no alignment, no site model or clock model needs to be specified. However, it still requires a tree prior, and this is what makes this analysis especially useful, since by carefully selecting an appropriate tree prior, we can answer questions of interest. In this case, the questions revolved around whether schisms in religions occur at a constant rate, or whether there are shifts in the rate.
The multitype birth–death model (Barido-Sottani et al, 2020) is especially well suited to infer rate shifts in phylogenies, and is implemented in the MSBD package for BEAST2. The model allows a prior to be set on the number of rate shifts. Nested sampling (Russel et al, 2019) was applied to perform hypothesis tests between having no rate shifts and having rate shifts, and Bayes factors estimated based on the marginal likelihood estimated from the nested sampling analysis.
It indicates there are no substantial rate changes in Indo-Iranian, one substantial change in Islamic and two substantial rate changes in Judeo-Christianity – one around the Lutherian reformation, and one shift in the Jewish branch. Below is the combined MCC trees for the three posterior samples of phylogenies obtained by nested sampling.
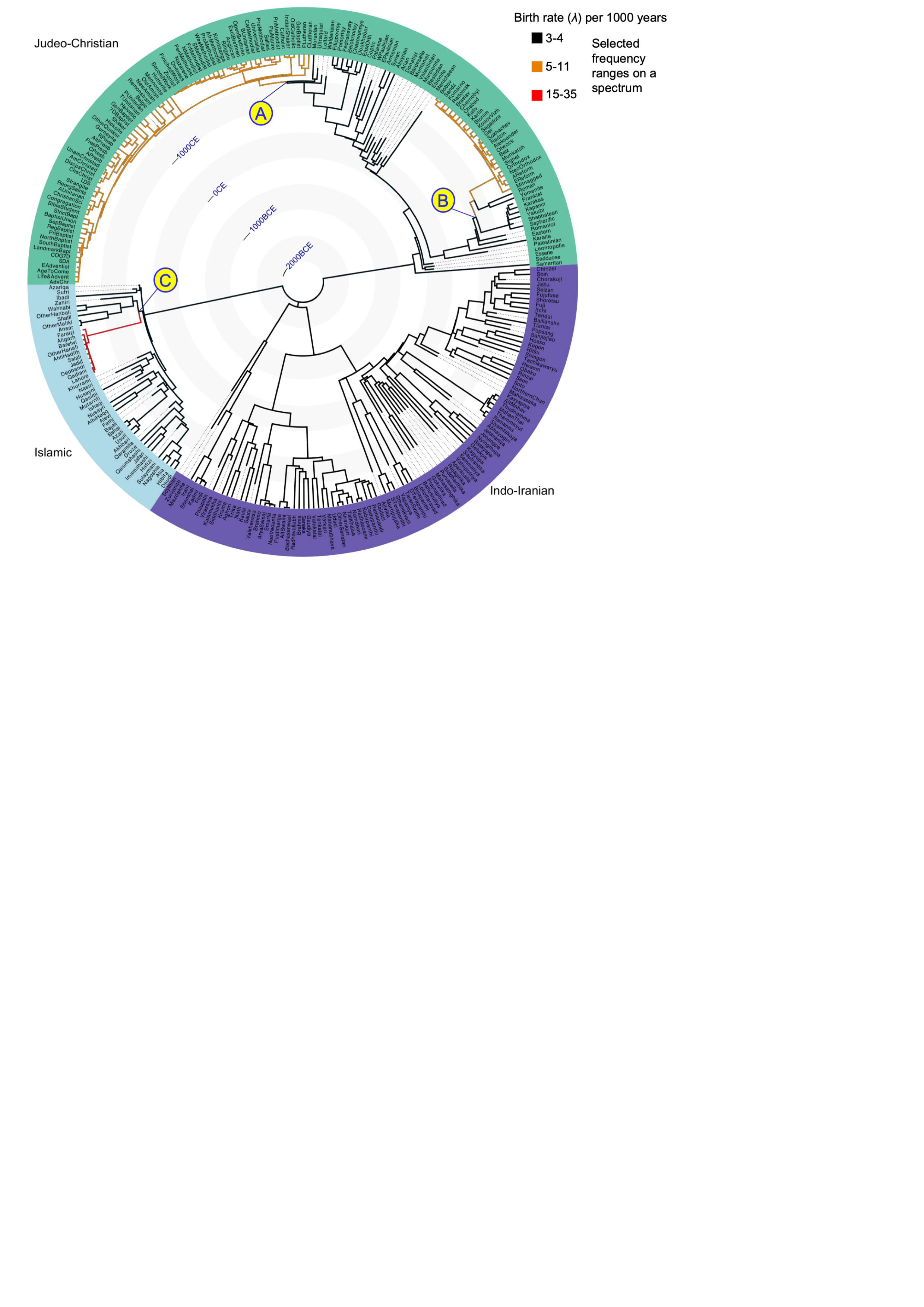
Here, the branches are colour-coded by mean birth rate and letters (A-C) mark the branches along which birth rate shifts were inferred. For finer resolution, see the Zenodo repository ‘Global tree figure’.
Though this is not a typical analysis, it does show that interesting results can be obtained using BEAST2 without sequence alignments.
References
Barido-Sottani J, Vaughan TG, Stadler T. A multitype birth–death model for Bayesian inference of lineage-specific birth and death rates. Systematic biology. 2020 Sep 1;69(5):973-86.
Anastasia Ejova, Oliver Sheehan, Simon J Greenhill, Jakub Cigán, Silvie Kotherová, Jan Krátký, Radek Kundt, Eva Kundtová Klocová, Joseph Watts, Remco Bouckaert, Quentin D Atkinson, Joseph Bulbulia, Russell D Gray. Three evolutionary radiations shaped the evolution of global religious diversity. Evolutionary Human Sciences, Volume 7, 2025, e43, doi:10.1017/ehs.2025.10027.
Russel PM, Brewer BJ, Klaere S, Bouckaert RR. Model selection and parameter inference in phylogenetics using nested sampling. Systematic biology. 2019 Mar 1;68(2):219-33.