31 March 2022 by Jordan Douglas
StarBeast3 is a BEAST 2 package for performing inference efficiently under the multispecies coalescent (MSC). Historically, researchers have often concatenated sequences of individual genes together into one long sequence and inferred the phylogeny of the species as that of the concatenated sequence. However, this approach is associated with numerous statistical pitfalls, including an overestimation of species divergence times. The MSC model corrects for incomplete lineage sorting and independency of gene trees, and is the next step beyond this traditional approach.
In this blog post we will describe the MSC model, how StarBeast3 gains efficiency over other software packages, and we provide a rough guide on how long it takes to achieve convergence. A tutorial describing how to use StarBeast3 and set up a StarBeast3 analysis in BEAUti can be found here.
The multispecies coalescent
Under the multispecies coalescent (MSC) model, one or more gene trees are constrained within a single species tree. The phylogeny of the species often differs from that of a gene. Consider the figure below, which depicts an MSC model based on Gopher data by Belfiore et al. 2008. Here, there are two gene trees (red and blue) constrained within a single species tree (black). There are 8 species (labelled at the bottom) and 27 individuals associated with each gene. Each gene tree is associated with a multiple sequence alignment, and each species is associated with a number of individuals from each alignment. The y-axis describes divergence time, in units of substitution per site. Shown here is just one of many states sampled in the posterior distribution.
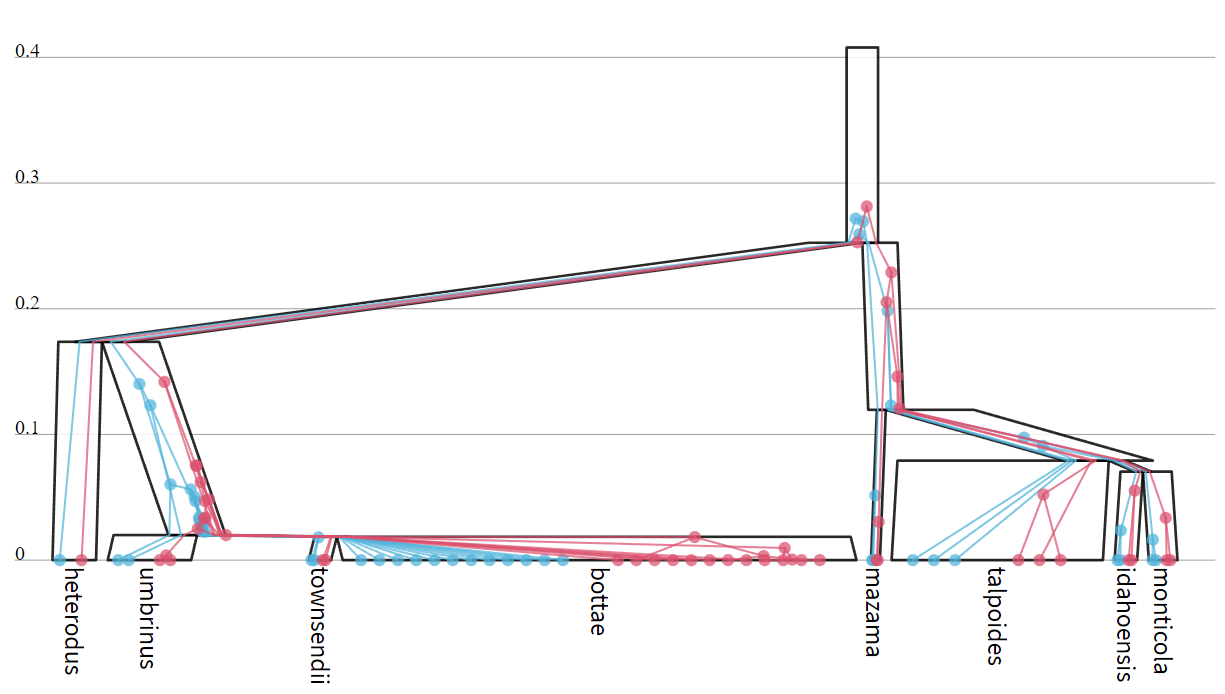
The width of each species branch above is proportional to the effective population size Ne of that lineage. Smaller values of Ne are associated with a faster rate of coalescence, reflecting lower genomic variation in the population. For example, consider the (Thomomys) bottae and T. monticola species above. The former has a large Ne (wide branch), reflecting most of the coalescent events (blue or red circles) occuring earlier in time before the origin of the species (in the lineage above [T. umbrinus, T. townsendii, T. bottae]). Whereas, the latter has a smaller Ne, reflecting its coalescent events occuring within the T. monticola species. In the case of Orthogeomys heterodus, there was only one individual sampled from the species, and therefore there is no direct signal informing its effective population size.
Parameters
In StarBeast3, the parameter space is quite large, and often includes:
- One species tree.
- One or more gene trees.
- One site model associated with each gene tree. A site model includes a substitution model (eg. HKY), a (relative) gene-tree substitution rate, a proportion of invariable sites, and optionally a gamma shape describing rate heterogeneity.
- One substitution rate associated with each branch of the species tree. If a strict clock model is assumed, then these rates are fixed at 1. But under a relaxed clock model, they are estimated. The rate of evolution in a gene tree branch is governed by that of the species tree branches which contain it.
- One effective population size Ne associated with each branch in the species tree. Under the prior distribution, these terms are independent and identically distributed under an inverse gamma distribution.
- The mean effective population size (for the inverse gamma distribution).
- Species tree prior parameters (e.g. for a Yule model).
To reduce the parameter space, gene trees or site models can optionally be linked (meaning that one gene tree / site model is associated with more than one gene sequence).
Advanced MCMC operators
The parameter space is clearly quite large, and efficient MCMC proposal kernels (aka operators) are needed to facilitate search in a timely manner. StarBeast3 was built on top of StarBeast2, and has introduced several additional operators to the MSC model to improve convergence. Some of these operators are detailed below:
Parallel gene tree operator
Gene trees (and their sites models) are assumed to be independently distributed (conditional on the species tree and its branch parameters). StarBeast3 exploits this assumption by parallelising the MCMC inference of gene trees. For example, if there are 100 gene trees, and the user allocates 4 threads to this operator, then a thread will operate on 25 gene trees in parallel when the operator is called. This operator saves time at operating on gene trees directly (but not when operating on the species and gene trees simultaneously).
Effective population size Gibbs operator
As a result of estimating the effective population sizes (instead of integrating the term out, as StarBEAST2 and STACEY both do), StarBeast3 employs a Gibbs operator to propose new terms for all effective population sizes simutaneously. This method is superior to operating on each Ne term one-at-a-time using a random walk operator, for example.
Constant distance operators
In the special case of the relaxed clock model, StarBeast3 extends the constant distance operators used in the ORC package. These operators move species tree divergence times, gene tree node divergence times, and species tree branch rates simultaneously, such that the evolutionary distance between all taxa remains constant. This operator is very efficient, and therefore makes the relaxed clock model an appealing choice for StarBeast3.
Bactrian kernel, AVMN kernel, and more
For further details on the operators used by StarBeast3, please see the main article.
Comparative benchmarking
We benchmarked StarBeast3 against StarBEAST2 – which was demonstrated to be an order of magnitude faster than starBEAST – for their abilities to achieve convergence in different areas of parameter space during MCMC. Different parameters were found to converge at quite different rates, however most notably, we found that the posterior density converged between 2 and 36 times as fast for StarBeast3, and relaxed clock branch rates converged between 3 and 35 times as fast. The discrepancy between StarBeast3 and 2 was most prominent for the larger datasets. On smaller datasets (e.g. < 30 genes), StarBeast3 and 2 are likely to show similar behaviour.
But how long will it take?
Every genomic dataset is different. They vary in the number of species, the number of indviduals per species, the number of genes, and the length of the genes. Some datasets are very clock-like and contain strong signal supporting a small field of similar topologies which are readily traversed. Others contain poor-signal or mixed-signal and produce multimodal distributions that take longer to traverse.
Some researchers may be wondering how extensively their dataset must be subsampled in order for StarBeast3 to be of use. So here we provide a very rough guide which we hope will be useful.
We ran 5 replicates of StarBeast3 across 5 datasets and recorded the range (min-max) of times required for the posterior density to converge (i.e. to achieve an effective sample size of 200, after discarding the first 20% of states). All were run under the relaxed clock model. This is what we found:
| Dataset | Number of species | Number of taxa | Number of gene trees | Time to convergence |
|---|---|---|---|---|
| Frog (Barrow et al. 2014) | 21 | 88 | 26 | 1-2 days |
| Skink (Bryson Jr et al. 2017) | 10 | 59 | 50 | 2-3 days |
| Spider (Hamilton et al. 2016) | 36 | 83 | 50 | 28-46 days |
| Simulated dataset 1 | 4 | 12 | 100 | 1-4 days |
| Simulated dataset 2 | 16 | 48 | 100 | 18-40 days |
We also showed that StarBeast3 is capable of processing up to 1000 gene trees, but on small trees (4 species and 12 taxa; strict clock model).
Predicting the convergence time of a dataset is a difficult task. However, as a starting point we recommend subsampling datasets down to fewer than 100 gene trees (e.g. 50), and then progressively adding more gene trees (e.g. 50, 75, 100, 150, 200, … ) until convergence is no longer feasible.
Useful links
Find StarBeast3 on GitHub BEAST users page
References
Douglas, Jordan, Cinthy L. Jimenez-Silva, and Remco Bouckaert. “StarBeast3: Adaptive Parallelized Bayesian Inference under the Multispecies Coalescent.” Systematic Biology (2022). DOI
Ogilvie, Huw A., Remco R. Bouckaert, and Alexei J. Drummond. “StarBEAST2 brings faster species tree inference and accurate estimates of substitution rates.” Molecular biology and evolution 34.8 (2017): 2101-2114. DOI