1 August 2022 by Jordan Douglas
Species delimitation is the task of defining boundaries between species - a problem which has persisted in biology for eons. There exist numerous rapid likelihood-based approaches to this problem which can define species boundaries from genomic data. However, these approaches are largely restricted to single-locus data and are thus unable to account for the fact that genomic regions frequently have different evolutionary histories from one another, and from the species itself. The coalescent-based species concept accounts for these processes and also enables the quantitative testing of different hypotheses. This approach relies on the multispecies coalescent (MSC) model.
In this post I will give a general overview of the newly released SPEciEs DEliMitatiON package (SPEEDEMON) for BEAST 2. This application performs fast Bayesian inference on both a) multilocus biological sequence data, and b) SNP data, all under the multispecies coalescent framework. SPEEDEMON is built on top of a complex hierarchy of phylogenetic methods, which have been previously implemented in the StarBeast 1-3, SNAPP/SNAPPER, DISSECT/STACEY, and BICEPS packages for BEAST 2.
A user-guide describing how to install and use SPEEDEMON can be found here.
The YSC tree prior
Species delimitation is implemented as a tree prior distribution known as the Yule-skyline collapse (YSC) prior. The model is the combination of two tree models:
-
The threshold/collapse model implemented in the DISSECT and STACEY packages for BEAST 2. Under this model, taxa whose divergence time falls below some small user-defined threshold ε are collapsed into the same species. The figure below shows the tree prior density under this model. Taxa who fall into the rectangular part of the density (the spike) are considered to be part of the same species, and those in the exponential curve (the slab) are regarded as separate species. The collapse weight ω controls the area under the spike. Figure from ref [1].
-
The Yule skyline model implemented in the BICEPS package. Under this model, ancestral speciation rates vary through time in a smooth piecewise fashion, similar to the Bayesian skyline model.
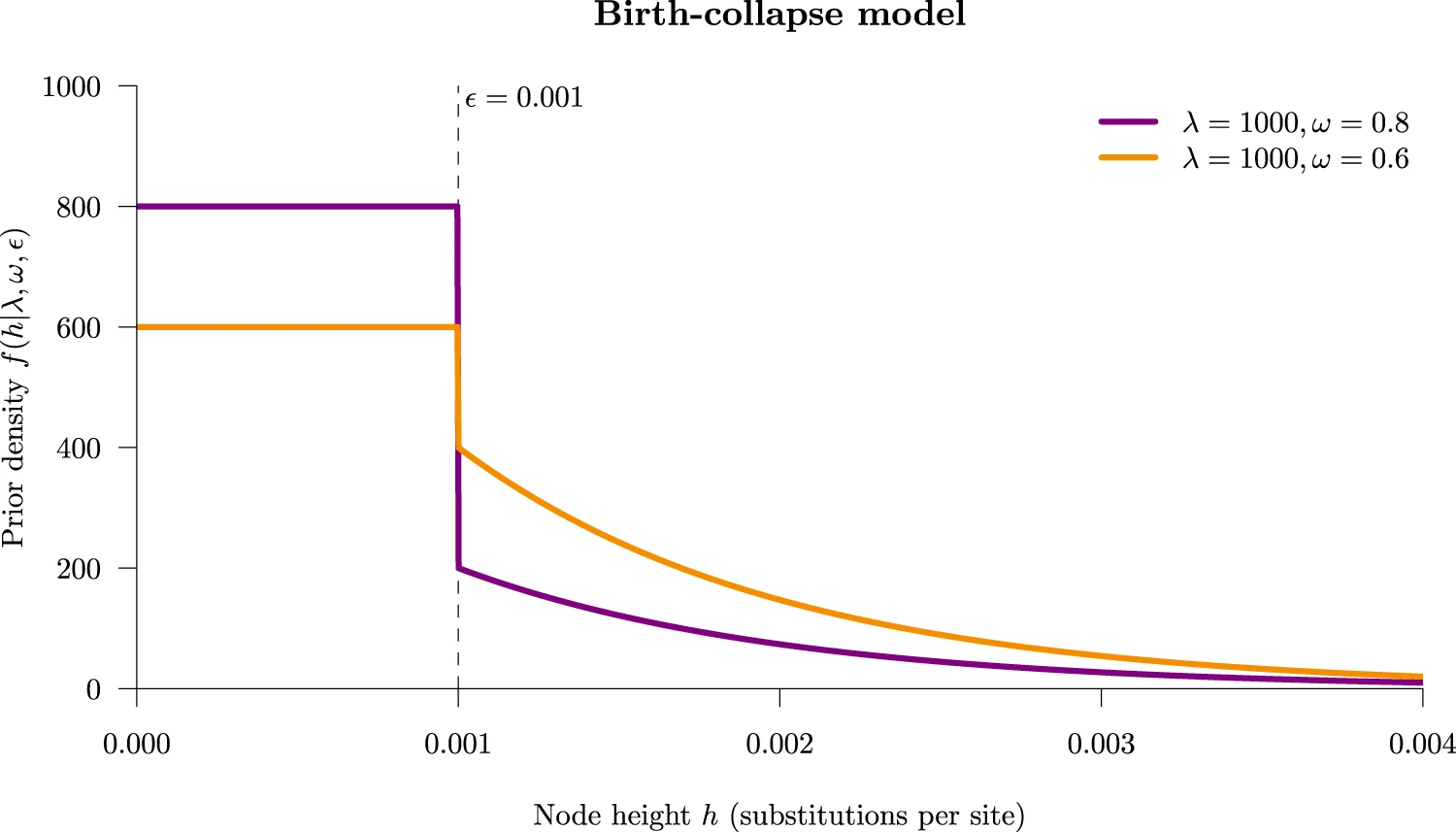
Taken together, the YSC model clusters taxa into a species if their divergence times are small, while also allowing the diversification rate to vary through time.
Data types
SPEEDEMON can work with SNP data as well as multilocus biological sequence data. These two data types are associated with different likelihoods and different BEAST 2 packages.
SNAPPER: SNP data
In this model, each SNP is modeled with its own phylogeny (which are integrated out) constrained within a species tree (which is estimated). The SNAPPER package for BEAST 2 has an accurate approximation that allows a fast implementation of this model. The YSC tree prior can be used in conjuction with SNAPPER to infer species boundaries from SNP data. I refer the reader to ref [1] for an application on Gecko SNP data.
StarBeast3: Multilocus sequence data
In this model, each biological sequence is associated with its own phylogeny (the gene trees), which are constrained within the species tree. Each gene tree is associated with its own substitution model, and each branch in the species tree is associated with an effective population size and a substitution rate, under the relaxed clock model. This is implemented in StarBeast3.
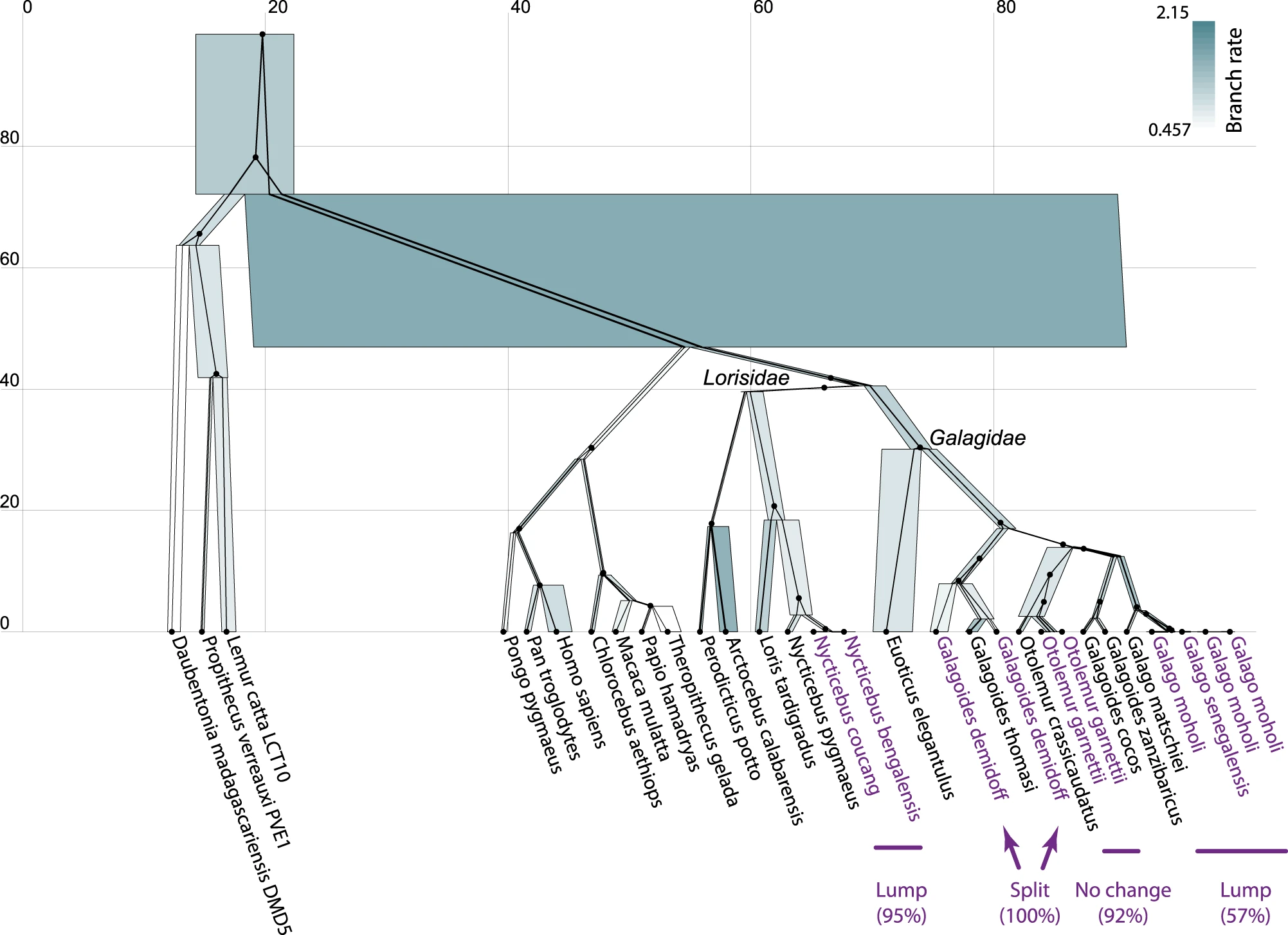
I refer the reader to ref [1] and its figure below for an application on multilocus primate data (Pozzi et al. 2014). Here, one of several gene trees are shown constrained within the species tree. Node heights are in units of millions-of-years. Species node widths are proportional to effective population sizes and are coloured by subsitution rate under the relaxed clock model. Taxa which were lumped into species by SPEEDEMON are indicated alongside their posterior support.
Selecting the threshold ε
The threshold ε describes the maximum divergence that can be tolerated before two samples are regarded as separate species. Too large, and all taxa will be lumped into one species. Too small, and each taxa will be split into their own species.
ε defaults to 1e-4 in BEAUti, however I strongly encourage users to explore a range of values of ε, rather than treating it as a black-box constant. I note that species delimitation appears to be quite robust to ε in general, but a sensitivity analysis is still important on a case-by-case basis.
I refer the reader to the guide in ref [1] for selecting a range of values for ε, which is summarised below:
- Run a preliminary phylogenetic analysis without SPEEDEMON to get a feel for divergence times (i.e. node heights). ε will later be expressed in the same units as the tree times. By default, this is expected number of substitutions per site, but if calibration data are used, then it could be years, or millions of years etc.
- If there are any two taxa which you know are not the same species, then use their divergence times to inform the upper bound of ε. This can also be informed by a related system (e.g. there is approximately 1% divergence between humans and chimps, so ε should be much less than 1e-2 substitutions per site in this system).
- If there are two taxa which you know are the same species, then use their divergence times to inform the lower bound of ε.
- Run multiple MCMC replicates exploring a range of thresholds ε between the lower and upper bound.
See also the review by Carstens et al. [6], who emphasise the importance of using multiple methods for species delimitation, and that species delimitation should be conservative (corresponding to larger values of ε).
Useful links
References
[1] SPEEDEMON: Jordan Douglas and Remco Bouckaert. Quantitatively defining species boundaries with more efficiency and more biological realism. Communications Biology 5, 755 (2022) DOI
[2] STACEY: Jones, Graham. Algorithmic improvements to species delimitation and phylogeny estimation under the multispecies coalescent. Journal of mathematical biology 74, no. 1 (2017): 447-467. DOI
[3] BICEPS: Bouckaert, Remco. An Efficient Coalescent Epoch Model for Bayesian Phylogenetic Inference. Systematic Biology (2022). DOI
[4] SNAPPER: Stoltz, Marnus, Boris Baeumer, Remco Bouckaert, Colin Fox, Gordon Hiscott, and David Bryant. “Bayesian inference of species trees using diffusion models.” Systematic Biology 70, no. 1 (2021): 145-161. DOI
[5] StarBeast3: Douglas, Jordan, Cinthy L. Jiménez-Silva, and Remco Bouckaert. “StarBeast3: Adaptive Parallelized Bayesian Inference under the Multispecies Coalescent.” Systematic Biology (2022). DOI
[6] Carstens, Bryan C., Tara A. Pelletier, Noah M. Reid, and Jordan D. Satler. “How to fail at species delimitation.” Molecular ecology 22, no. 17 (2013): 4369-4383. DOI