1 September 2024 by Remco Bouckaert
Introduction
In a recent paper (Ferretti et al, 2024) it was demonstrated that when sequence data on a phylogeny is generated with site rates independently drawn from a continuous gamma distribution, the commonly used discretised gamma rate heterogeneity model using 4 categories (Yang, 1996) over estimates branch lengths, while the free-rate model (Soubrier et al, 2012) does not suffer from the same problem.
Conceptually, the free rate model is more satisfying, since before performing a distribution it is usually unknown which site rate distribution generated the data at hand, and the free rate model does not commit to a parametric distribution. Also, the discretised gamma model depends on implementation details (e.g., use mean or median of categories, and even the implementation of the gamma function) that can lead to substantially different outcomes in an analysis. So, analyses between different software implementations may be incomparable.
This raises the question, should we abandon the discretised gamma rate heterogeneity model and switch to the free rate model where possible? Here, we consider this question in a Bayesian setting. There are no computational penalties for switching, but where the gamma rate heterogeneity model has only a single parameter (the gamma shape parameter determining the shape of the gamma distribution), an equivalent free-rate model with 4 categories has 6 parameters: 3 for weights and 3 for rates.
Free rate model in BEAST2
The free rate model is implemented in BEAST2 in the RBS package. In the free rate model with \(k\) categories, let \(w_i\) be the weight for \(i=1,\ldots,k\) and let \(r_i\) be the rate. All weights and rates are positive real numbers and weights are constrained to sum to 1 (\(\sum_{i=1}^kw_i=1\)) and mean weighted rates are 1 as well (\(\sum_{i=1}^kw_ir_i=1\)). For ease of inference, we use a Dirichlet prior on weights, thus guaranteeing they sum to 1. Further, instead of sampling rates \(r_i\) directly, we sample parameters \(x_i\) independently from a Dirichlet distribution, so they sum to 1 (\(\sum_{i=1}^kx_i=1\)). Next, we set \(y_i=\sum_{j<=i}x_i\), thus guaranteeing \(y_i\le y_{i+1}\), which helps identifying rates. Finally, we set the rates \(r_i\) proportional to \(y_i\) with a scale factor to guarantee that \(\sum_{i=1}^kw_ir_i=1\). The scale factor is simply \(\frac{1}{\sum_{i=1}^kw_iy_i}\).
This parameterisation probably differs slightly from that described in Soubrier et al. (2012), since the description in the paper was not sufficiently detailed to implement. MCMC proposals for weights as well as for rates are the default delta exchange operators in BEAST2.
To verify that the model is correctly implemented, we performed a well calibrated simulation study over trees with 50 taxa drawn from a Yule prior with birth rate log-normally distribution with \(M=0.5, S=0.1\). For the substitution model, we used the HKY model with frequencies distributed according to a Dirichlet(4,4,4,4) distribution and kappa according to a log-normal distribution with \(M=2, S=1.25\). We used a strict clock with tight normal prior on rates with mean 0.1 and stand deviation 0.005. This together with birth rate prior provides the timing information for the trees. The range of tree heights was between 5 and 16. For the free rate model, 4 categories were used with where we used a Dirichlet(4,4,4,4) for weights and Dirichlet(1,1,1,1) for rates.
We drew 100 instances from the above model and generated sequences of size 100, 250, and 500. Then, we performed MCMC analyses for each of these instances. We tested how often the 95% highest probability intervals (HPD) covered the “true” value, that is the value used to generate the sequence data. If the 95% HPDs contains the true value between 91 and 99 out of the 100 experiments (for each of the three sequence lengths) we deem the inference accurate. Coverage for tree height, tree length, kappa, frequencies, birth and clock rate all fell within the desired range, so we find that the implementation in BEAST2 can infer phylogenies well and probably does not contain major flaws.
Is tree height overestimation a problem for Bayesian inference?
To confirm the results of Ferretti et al. (2024), we performed a variant of the well calibrated simulation study outlined above where we generated alignments, but instead of drawing from four rate categories for each site individually we draw a rate from a continuous gamma distribution with shape of 0.5 (following Ferretti et al. 2024). Then, we infer the tree under the discretised gamma rate heterogeneity model (with shape distributed according to an exponential distribution with mean 1) and under the free rate model (with priors outlined above). Again, we draw 100 alignments with 100, 250 and 500 sites per alignment over a tree with 50 taxa.
Where the tree height and length coverage for the free rate model is within expected range, the coverage for the discretised gamma rate heterogeneity model was only 85, 75 and 70 for tree length for the three increasing site lengths respectively. For tree height it was 92, 87, and 82 respectively, so only the smaller alignments passed the test. Inspecting the coverage plots shows that tree heights and lengths tend to be over-estimated, even though the original rates draw from a (continuous) gamma distribution. So, the effect demonstrated by Ferretti et al. (2024) in a maximum likelihood setting is present in a Bayesian setting as well: the free rate model can absorb site rate variation caused by long tails of the gamma distribution while the discretised gamma model does not.
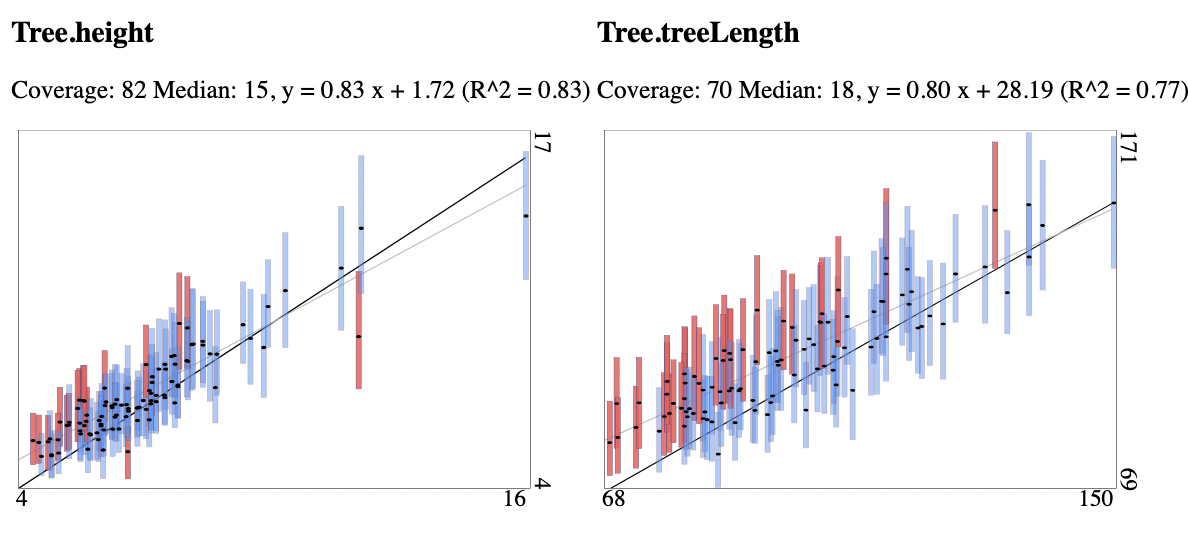
Fig. 1 95% HPDs for each of the 100 runs for alignments with 500 sites. True tree height and length on the x-axis, estimates on the y-axis. When the true value is covered by the 95% HPD, the bar is blue, otherwise it is red. Alignments generated with sites drawn from continuous gamma rate distribution and estimas based on 4 category discretised gamma rate heterogeneity model. The model overestimates tree heights and lengths.
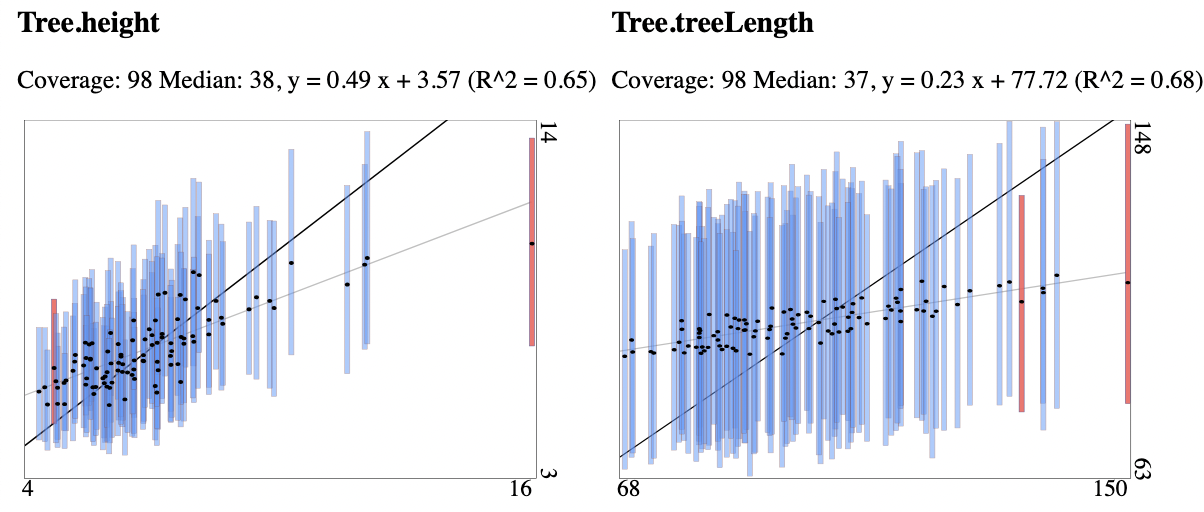
Fig. 2 As Fig. 1, but estimates based on free rate model. The model correctly estimates tree heights and lengths.
So, should we always use the free rate model?
The free rate model has more parameters, and usually the site model parameters have sufficient data available for these parameters to be estimated quite accurately. For example, there are 50 times 500 four valued data points, that is 50 thousand bits of information in our simulation studies. However, since the free rate model affects the scaling of the tree, it injects quite a bit of uncertainty in the over-all tree height. Since the weights can vary, the associated rates can compensate and it looks like this has a substantial effect on tree height and length estimates. For example, the average size of the 95% HPD for the free rate model for the 500 site alignments is 4.36 for tree height and 56.5 for tree length, while for the discretised gamma model it is just 2.52 and 28.0 respectively.
So, uncertainty is increased by about a factor two by the free rate model. In other words, the free rate model reduces bias, but increases variance.
Can uncertainty be reduced in the free rate model?
One way to reduce the uncertainty is by fixing the weights to be equal \(1/k\), so 0.25 for four categories. This allows less flexibility for the values of the rates, and thus reduces uncertainty in tree age and length estimates. Unfortunately, repeating the simulation study under this condition makes the coverage for tree height fall outside the expected range (just 87, 77, 76 for the 100, 250 and 500 site sequences respectively). This confirms that it is the static category weights that cause the bias, as implied by Ferretti et al.
Alternatively, the prior on weights can be tightened: when changing it to a Dirichlet(20,20,20,20), for 500 site sequences, coverage of tree height is 91 and 95 of tree length, so within expected range. Furthermore, the average 95% HPD for tree height reduces to 3.36 and for tree length to 43.2.
With a Dirichlet(100,100,100,100) coverage becomes 84 for tree height and 80 for tree length, which is not acceptable, and an average tree height 95% HPD of 2.8 and tree length HPD of 32.8, so only slightly more uncertain than that of the discretised. A Dirichlet(50,50,50,50) gives coverage 87 for tree height and 87 for tree length, 95% HPD widths of 2.97 and 36.5. However, a Dirichlet(30,30,30,30) gives coverage 91 for tree height and 92 for tree length, so within expected range, but still an average tree height 95% HPD of 3.19 and tree length HPD of 40.3, so still elevated from the discretised gamma model, though reduced from that of a less tight weight prior.
Increasing the number of categories to 8 while fixing all category weights to 1/8 gives a coverage of 95 and 92 and mean 95% HPDs of 2.33 and 26.8 for tree height and length respectively. The uncertainty is slightly reduced from the 4 category discretised gamma model.
Can bias be reduced in the discretised gamma rate model?
One potential solution is to use more than the commonly used four rate categories in the discretised gamma rate heterogeneity model. We repeated the simulation study with 8 categories instead of 4. Doubling the number of categories implied doubling the calculation time. This increased the coverage of tree height to 94, 98 and 90 for the 100, 250 and 500 site sequences respectively. Likewise, for tree length the numbers are 93, 95 and 93 respectively. So, adding rate categories makes the problem go away, keeps the uncertainty of the root age and tree length estimates under control, but at the cost of extra calculation time.
For 500 sites, the average 95% HPD size is 2.54 for tree height and 28.9 for tree length, so not substantially different from the 4 category discretised gamma model.
Are clade support estimates affected by the choice of model?
The average entropy according to the conditional clade distribution (CCD1) is 9.35 for the 8 category gamma model for 500 site sequences and 9.36 for the 8 category free rate model, so practically indistinguishable.
Also, clade coverage tests in all instances showed nice triangle plots, suggesting posterior clade support estimates are accurate and unbiased for all models and sequence lengths, and the number of clades with a certain support (i.e. size of bins in triangle plots) does not differ substantially.
It appears that biases only affect age estimates.
Conclusions
The observation from Ferretti et al. (2024) that the 4 category discretised gamma rate heterogeneity model is biased in age estimates while the free rates model is not for simulated data with site rates from a continuous gamma distribution is confirmed in a Bayesian setting. However, this holds only when free rate model weights are estimated, and estimating weights injects substantial uncertainty in age estimates.
There does not appear to be much difference between discretised gamma and free rate model when fixing the category weights for simulated data with site rates from a continuous gamma distribution: when using 4 categories, both methods fail, but when using 8 categories both methods pass the test.
What can be taken from these observations is that it makes sense for your data to check whether estimates are sensitive to the number of rate categories, that is, run an analysis with 4 categories, and repeat with 8 categories and compare the results. If there are substantial differences, trust the 8 category model results over the 4 category model.
It pleads for the free rate model (with fixed weights) that it is a non-parametric model that allows for a wider range of distributions than the continuous gamma distribution.
Free rate model availability
To use the free rate model in BEAST2, install the RBS package.
In BEAUti, go the Site model panel and select “Free rate model” from the drop-down box at the top of the screen.
If you want to estimate weights (not recommended), select the estimate check box next to the weights entry.
Be careful when adding more categories: default priors are Dirichlet priors and when adding categories the parameters of Dirichlet should be updated in the Priors panel to have matching numbers of parameters.
References
Ferretti L, Golubchik T, Di Lauro F, Ghafari M, Villabona-Arenas J, Atkins KE, Fraser C, Hall MD. Biased estimates of phylogenetic branch lengths resulting from the discretised Gamma model of site rate heterogeneity. bioRxiv. 2024:2024-08.
Soubrier J, Steel M, Lee MS, Der Sarkissian C, Guindon S, Ho SY, Cooper A. The influence of rate heterogeneity among sites on the time dependence of molecular rates. Molecular biology and evolution. 2012 Nov 1;29(11):3345-58. doi:10.1093/molbev/mss140
Yang Z. Among-site rate variation and its impact on phylogenetic analyses. Trends in ecology & evolution. 1996 Sep 1;11(9):367-72.